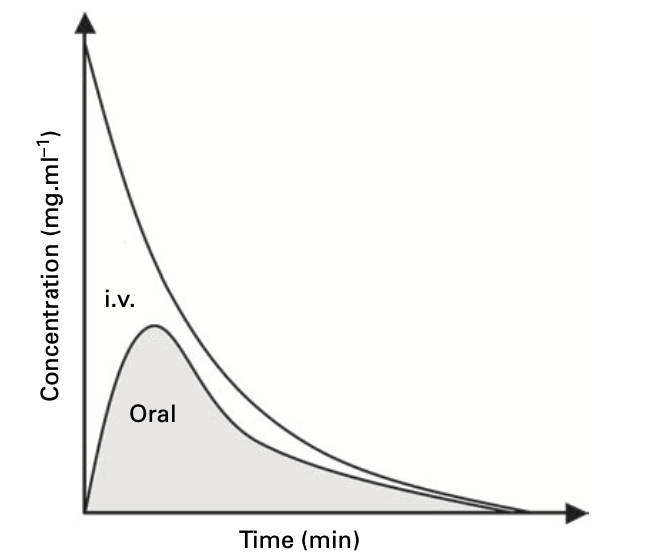
- The extraction ratio is the fraction of a drug removed from the blood by an organ with each pass through that organ
- For example, for an oral drug:
Bioavailable fraction = Fraction absorbed x fraction remaining after gut and hepatic metabolism
FB = FA x FG x FH
- Hepatic extraction ratio depends on:
- Hepatic blood flow
- Hepatocyte uptake of the drug
- Metabolic capacity of the hepatocyte for the drug (dependent on the Michaelis constant of the enzyme, the [substrate] at which the enzyme is working at 50% maximal rate)
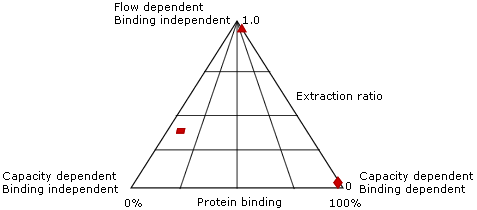
- Drugs typically fall into three groups:
- Flow dependent, protein binding independent
- Capacity dependent, protein binding dependent
- Capacity independent, protein binding independent
1 - High extraction ratio; high metabolic capacity, rapid hepatocyte uptake
- E.g. propofol, lidocaine
- The free drug is rapidly removed from plasma
- Therefore protein-bound drug is released
- The drug is rapidly metabolised within the hepatocyte
- This establishes a concentration gradient between the plasma and hepatocyte
- The drug's metabolism and extraction ratio is therefore highly dependent on hepatic blood flow and independent of protein binding
2 - High (>90% protein binding), low metabolic capacity
- E.g. phenytoin, diazepam
- The metabolism is limited by the metabolic capacity of the hepatocyte
- If protein binding is altered, then the [free drug] increases
- Initially, there is increased hepatocyte uptake but unchanged metabolism
- Once enzymes are saturated, intracellular levels increase and the concentration gradient disappears
- Hence the plasma free concentration rises and may cause side effects
- The drug's metabolism and extraction ratio is therefore influenced by metabolic capacity and protein binding, not hepatic blood flow
3 - Low protein binding, low metabolic capacity
- The total amount of drug metabolised is unaffected by hepatic blood flow or protein binding, and is largely dependent on enzymatic capacity to metabolise the drug
- Absorption takes place through gut mucosa, and is therefore impacted by gut motility and GI pathology
- Only unionised drugs and those with specific transport membranes make it through the lipid membranes of the gut
- The pH of the gut varies from stomach (acidic pH, acidic drugs unionised) through to duodenum (alkali pH, basic drugs unionised)
- There is variable absorption of drugs at different sites
- The salts of permanently charged drugs e.g. vecuronium, glycopyrrolate are not absorbed from the GIT
- In practice most drugs are absorbed in the small intestine due to its high surface area
- In general, oral route has the lowest bioavailability
Sublingual, nasal and buccal
- Rapid onset routes due to absorption through mucous membrane
- Higher bioavailability vs. oral as avoid first pass metabolism in the portal tract
- E.g. GTN, nifedipine SL, buccal midazolam
- Higher availability vs. oral as avoid first pass metabolism in the portal tract
- Small surface area vs. GI tract means slower absorption
- Considered for local effects (e.g. steroids in IBD) or systemic effects (diclofenac PR)
- Faster speed of onset than oral
- Bioavailable fraction approaches 1.0 as avoids problems associated with oral route
- Absorption depends on local perfusion
- Delayed absorption following IM administration:
- Reduces drug efficacy and multiple doses may therefore be given
- When perfusion is restored, there may be a sudden rise in plasma concentrations to toxic levels
- Administration may cause pain, haematoma or abscess
- Certain drugs are well absorbed sub-cut. e.g. LMWH
- May be indicated if compliance is an issue
- Absorption depends on local perfusion and a similar issue as with IM administration may occur
- E.g. subcutaneous insulin in the critically unwell patient
- Can be used for topical effect e.g. steroids, local anaesthetic
- Can be used to avoid first-pass metabolism and therefore increase bioavailability e.g. fentanyl, nitrates
- Highly lipid soluble drugs favour transdermal absorption e.g. fentanyl
- Absorption depends on local perfusion
- Can use patches to create a smooth pharmacokinetic profile i.e. slow, constant release of drug
- Iontophoresis is a special type of transdermal application whereby an electromotive force is used to drive medication through the stratum corneum of the skin
- It is used therapeutically e.g. in hyperhidrosis
- It is used diagnostically e.g in CF; pilocarpine iontophoresis is used to stimulate sweat glands
- Can be used for local effect e.g. bronchodilators, inhaled nitric oxide, adrenaline
- Can be used for systemic effect e.g. volatile agents have site of action in the CNS
- The large surface area for absorption can lead to rapid rises in systemic concentration and rapid onset of action at distant effector sites
- The particle size and method of administration determine whether a drug reaches just the upper airways or alveolus too; only droplets <1μm in diameter reach the alveolus
- Used to provide regional anaesthesia and analgesia
- E.g. LA, opioids, ketamine, clonidine
- Speed of onset of LA determined by portion of unionised drug i.e. pKa
- E.g. lidocaine has faster onset than bupivacaine
- Addition of NaHCO3 will increase local pH, increasing the unionised fraction and reducing onset time
- Significant amounts may be absorbed into systemic circulation, causing side effects e.g. LA toxicity, opioids
- Amount of drug required is less than epidural route
- Little reaches systemic circulation and therefore rarely causes direct drug side-effects
- Speed of onset determined by:
- Volume used
- Speed of injection
- Type of solution and positioning (e.g. use of heavy bupivacaine)