Morphine is a naturally occurring phenanthrene derivative
It is the reference opioid to which all others are compared and, like all opioids, it is a weak base
Presentation
Tablets and capsules e.g. sevredol
Suspensions e.g. oramorph
Available as both immediate-release and modified-release (e.g. zomorph) preparation
Suppositories
Solution for intravenous, intramuscular and subcutaneous use
Preservative-free solutions are available for epidural and intrathecal use
Varying strengths: 1mg/ml, 10mg/ml and 15-30mg/ml
Dosing
The usual quip is that the dose of morphine is 'titrate to effect' i.e. there is a high inter-individual variability in efficacy based on patient and pain factors
For PO dosing, a dose of 5mg - 30mg 4hrly is a reasonable opening gambit
For IV and IM use, a starting dose of 0.1 - 0.2mg/kg titrated to effect is reasonable (NB IV dose approximates to 2x PO dose)
SC use is generally avoided due to low lipid solubility
Neuraxial dosing:
Intrathecal: 100micrograms (although doses up to 1mg are reported)
Epidural: 2-3mg
Neuraxial dose can cause delayed respiratory depression due to poor lipid solubility
Used to treat moderate-severe acute nociceptive pain
Modified release preparations no longer recommended for management of acute pain
Not recommended for use in chronic neuropathic pain syndromes, although often patients are in receipt of it anyway
Molecular weight: 337
Aqueous solubility: 1 in 5,000μg/ml
Lipid solubility (logP at pH 7.4): -0.1
Relative lipid solubility: 1 (as it is the reference molecule)
Low skin flux
Absorption
pKa 8.0
At pH 7.4 it is only 23% unionised
When administered orally:
It is mostly ionised at gastric pH and therefore not absorbed
In the alkaline environment of the small bowel it is mostly unionised
Oral bioavailability 25-30% due to hepatic 1st pass metabolism
Distribution
Plasma protein binding 35%
Volume of distribution 3.5L/kg
Elderly patients have increased peak plasma levels due to reduced volume of distribution
Peak effect:
IV: 10mins
IM: 30 mins
Duration of effect: 3-4hrs
Brain concentration falls slowly due to poor lipid solubility and therefore plasma concentration of drug doesn't correlate will with effects
Metabolism
Metabolised mainly in liver, but also kidneys
Neonates are more susceptible to morphine's effects due to reduced hepatic conjugating ability
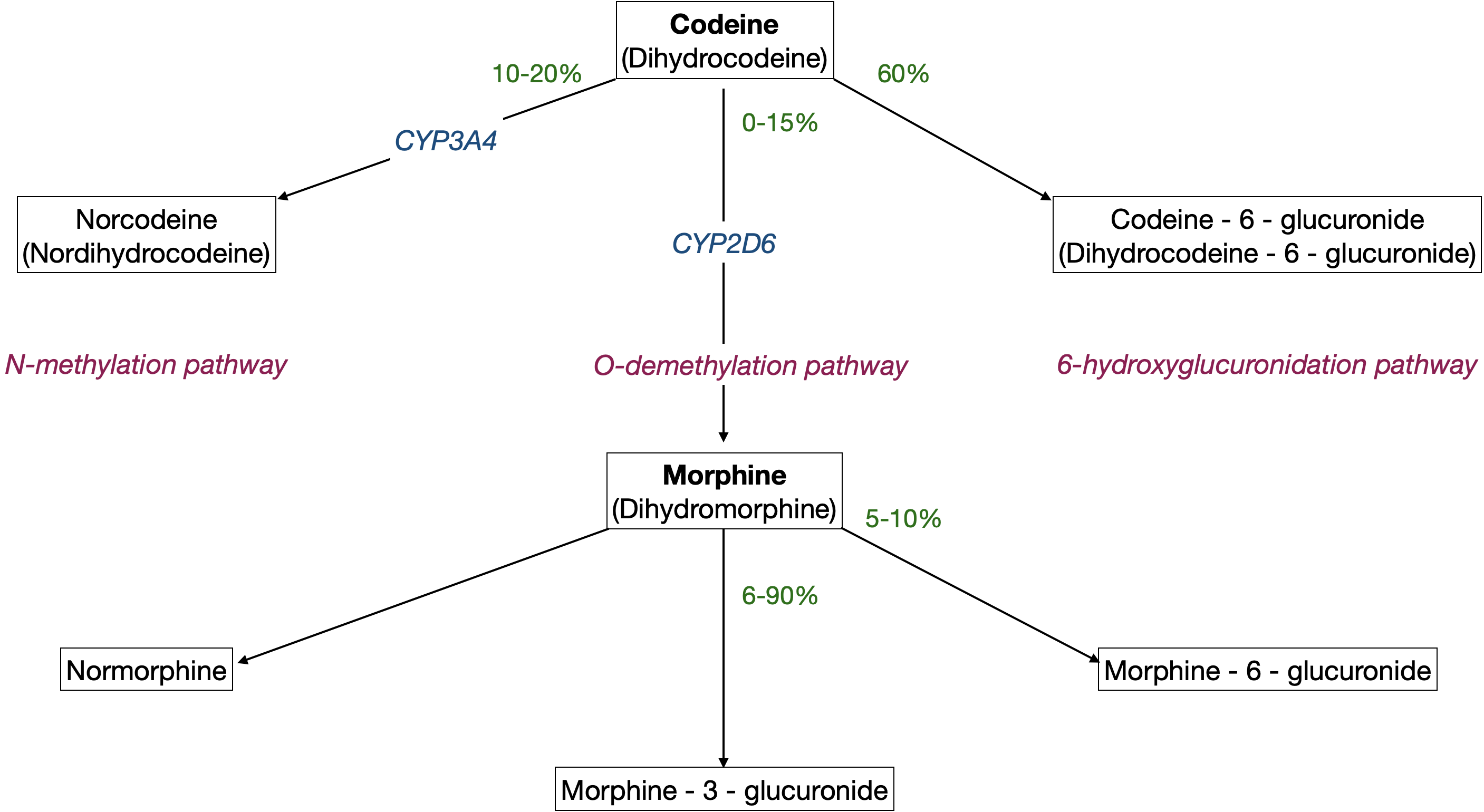
Mostly metabolised to morphine-3-glucuronide
Has effects on arousal
Possible MOP receptor antagonist
Excreted in the urine and accumulates in renal failure
Up to 10% metabolised to morphine-6-glucuronide
It's an active metabolite 13x more potent than morphine with a similar duration of action
Excreted in the urine and accumulates in renal failure
Some morphine is methylated to codeine, and it also undergoes oxidation, demethylation and conjugation with glucuronide
Excretion
Elimination half-life 170min
Clearance 16ml/min/kg
Respiratory
Reduces chemoreceptor sensitivity to CO2 although response to hypoxia is unaffected
If hypoxic stimulus is removed (e.g. removing supplemental oxygen) then respiratory depression may be potentiated
Reduces respiratory rate (to a greater extent than VT)
May precipitate bronchospasm through histamine release (dose-dependent)
Anti-tussive
Chest wall rigidity due to opioid receptor interaction with dopaminergic and GABAergic pathways in the substantia nigra and striatum
Cardiovascular
Reduces sympathetic tone, causing mild bradycardia and hypotension
Histamine release (dose-dependent) may also contribute to hypotension and cause peripheral vasodilation
No direct myocardial depressant effect
Neurological
Predominant MOP receptor agonism but also KOP and DOP receptor effects
Analgesia
Sedation, euphoria and then dysphoria with increasing doses
Meiosis - due to stimulation of Edinger-Westphal nucleus (treatment is atropine)
Renal
Causes urinary retention via:
Increased detrusor muscle tone
Increased ureteric tone
Increased vesical sphincter tone
Not nephrotoxic but active metabolites are renally excreted so can accumulate in renal impairment and lead to toxicity
Gastrointestinal
Constricts sphincters of the gut including the sphincter of Oddi
Inhibits Ach release by the myenteric plexus following MOP receptor agonism, leading to spastic immobility of the gut
Nausea and vomiting
Stimulates chemoreceptor trigger zone via 5-HT3 and D2 receptors
Suppresses cells of the vomiting centre
Dermatological
Rash and pruritus secondary to histamine release, reversed by naloxone
ruritus following IT/epidural administration is not secondary to histamine release nor association with rash, though may be helped by anti-histamines due to their sedating effects
Endocrine
Inhibits release of ACTH, prolactin and gonadotrophic hormone
Increases ADH secretion - may cause impaired water secretion and hyponatraemia
Semi-synthetic mixture of the anhydrous chlorides of the alkaloids of opium
Contains morphine (70%), codeine and papaverine
The component noscapine was removed from the formulation after it was found to be teratogenic in animal studies
Not given via IT or epidural routes due to preservatives
Effects are essentially the same as morphine's and it is reversed by naloxone